


FDA Q&A Regarding COVID-19 In-Vitro Diagnostic Tests
Introduction
On April 1, 2020 the FDA continued their Virtual Town Hall series to allow manufacturers and developers of COVID-19 tests the opportunity to ask questions to the FDA directly regarding the FDA Guidance on Emergency Use Authorization (EUA) and Policies on for In Vitro Diagnostic (IVD) Tests for Coronavirus Disease-2019 (COVID-19) during the Public Health Emergency.
In addition to the FDA policy for diagnostic test kits for COVID-19 issued on February 29, 2020, the FDA has issued an update on March 6, 2020 to include Policy A, B, C, and D for Coronavirus Disease-2019 (COVID-19) tests.
This call with the FDA included an extensive questions and answers panel open to the public to answer any questions. We have summarized the highlights of the call below to make it easier for you to learn the relevant information that was discussed.
Disclaimer: Please note this information may be paraphrased from actual statements, and do not represent actual verbatim statements from the FDA or callers.
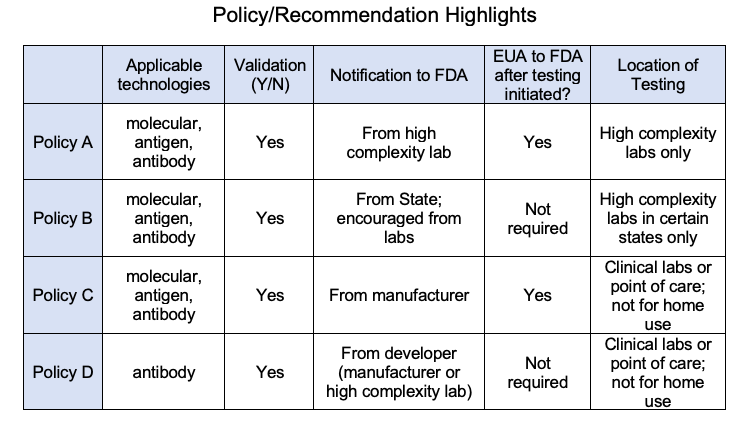
Source:
FDA Virtual Town Hall Series: Immediately in Effect Guidance on COVID-19
Diagnostic Tests March 25, 2020 (https://www.fda.gov/media/136439/download)
Do I need to submit for Emergency Use Authorization (EUA) for my laboratory IVD test which is a modification of an authorized test?
· The FDA does not intend to object to use of a test without a new or amended EUA; however, they expect labs to perform the appropriate validation under a bridging study.
Does data from validation tests or bridging tests need to be submitted to the FDA?
· Antibody IVD test kit developers under Policy D cannot claim authorization if they have not received EUA from the FDA. The FDA strongly encourages developers to perform a verification of the test performance; however this is not currently required.
· The FDA does not ask for the bridging testing to be submitted; however, but if you wish to share the information and share it voluntarily the FDA would like to see the data to put it on the FAQ page to help all labs.
Under Policy D, if a serological IVD test is validated for full blood, can it be used for capillary blood?
· Capillary blood can perform differently than whole blood. If the manufacturer’s instructions for use state that the sample is whole blood and the validation was performed only on full blood, the FDA suggests doing a bridging study between whole blood and capillary blood. The results of a bridging study are not required to be submitted to the FDA for review; however, the FDA is interested in reviewing this if a manufacturer volunteers to submit this data.
If my company has an antibody serological test that has been approved in the EU for some time, what do I need to do to sell it on the U.S. market?
· Notify the FDA and label it appropriately in accordance with Policy D. If you have CE mark for the IVD test kit, you may have enough data to get Emergency Use Authorization (EUA).
If one or more manufacturer or developers of antibody tests are interested in participating in a clinical trial, is there a contact office or person at the FDA they can talk to?
· FDA welcomes such studies and is happy to offer feedback. The FDA noted that they would expect to see a minimum of 30 positive patients with some IgM alone, some IgM/IgG, and ideally some IgG only. The FDA understands that given the current circumstances, the number of patients with only IgG is limited in the U.S., so they would expect it would be difficult for 30 IgG participants to be included.
· You can contact FDA at their email address to CDRH-EUA-Templates@fda.hhs.gov for further guidance.
With the serology tests, does the FDA have a source of SARS-CoV-2 positive material that IVD manufacturers could acquire for validation purposes. Is there any restriction in importing this positive material for validation?
· The FDA noted that there are overseas companies for this, but the manufacturer should assess the quality of this material. If manufacturers have trouble importing these materials, they should email the FDA.
· Additionally, the FDA is working with other agencies to help facilitate the collection and panels of immune serum for SARS-CoV-2 in the U.S. The FDA will update the FAQ page with regard to resources that could be used.
Does the FDA have any data on acceptability of saliva (nucleic acid) samples for detecting SARS-CoV-2?
· The FDA does not have policies currently issued for saliva nucleic acid tests for the detection of SARS-CoV-2 (COVID-19); however, they are interested in these tests.
· The FDA has reviewed test results for a tongue swab used in a clinic setting and the results were not promising. The FDA felt the tongue swab may not have provided enough saliva to yield promising results based on the data they have received and reviewed
· The FDA would want developers planning to sell a saliva nucleic acid test to submit a EUA amendment with the data before the FDA may authorize that test.
Considering EUA is not necessary for antibody testing under IVD Policy D as long as it is labeled appropriately, should manufacturers label their test kit for IVD use?
· Yes, labeling the test for IVD use is appropriate because these kits are being allowed to be marketed as IVDs under Policy D.
If I make minor updates to the labeling of my COVID-19 IVD test that falls under Policy D, do I need to notify the FDA of these changes?
· For minor changes to the labeling (e.g. extending the shelf life based), the FDA does not need to be notified. If there are changes to the performance, the FDA should be notified of the change.
What if I have a test that I want to sell for at-home use?
· The current EUA Policies for COVID-19 IVD tests do not apply to home use tests. The FDA requires IVD developers to contact the FDA to seek EUA authorization for at-home use.
· For in-home settings, the FDA is focusing on user safety, including safety of using any collection device, safety of the materials in the test device, and ensuring that results are correctly interpreted and linked back into the healthcare system. For these reasons, the FDA noted that a healthcare professional must be involved in some way to supervise the at-home testing and interpretation of the results.
· At-home use of serological tests introduces additional variables, and it is important that developers are able to demonstrate safety in these at-home environments. Blood sample collection, which is required for serological tests, is not performed routinely by lay users except for diabetics. The FDA acknowledges that the display of the results of the test devices are typically such that a healthcare provider can understand with little consideration for at-home use.
· The FDA suggested that there are clinician observation methodologies of a patient in the home that could be leveraged.
· The FDA recommends performing an observational study to identify if lay users (patients and caregivers) can use the device and instructions (under direct observation from a healthcare provider) to safely perform this testing at-home.
· The FDA also urges developers to consider how the patient will interpret the results, and how to have a clinician involved in this process. The FDA noted that some telemedicine portals are being proposed to have the user perform the test under supervision of healthcare providers to ensure the test done appropriately and results can be interpreted by a healthcare provider.
· Note: If you are looking to get your COVID-19 IVD approved for at-home use, please contact us to see how UserWise can help. We are experts in identifying use-related risks, preparing justifications for limited or remote usability testing, executing usability tests, and more (userwise@userwiseconsulting.com).
Would the FDA agree to allow for at-home use of a nasopharyngeal test? Would this need to be observed via telemedicine?
· The FDA expressed skepticism if a nasopharyngeal test could be self-administered. The FDA believes there is danger of self-harm or harm incurred by others performing this test on patients because of the invasive nature of the test. Additionally, the FDA is concerned about others helping patients collect this sample at-home because this means of collection could make patients cough or sneeze, which may spread the virus.
· Successful administration of anterior nasal swabs for nasopharyngeal tests have been demonstrated to be an effective test; however, the FDA would want to see data to prove that if a sample collected at home is returned for processing of the results, the sample is going to be in-tact and not produce false-negatives. The FDA noted that using a transportation medium that is already FDA cleared would likely not require additional testing. Otherwise, the FDA recommends performing benchtop studies (e.g. simulated shipping tests for temperature, etc.)
· Note: Foam swabs for anterior nasopharyngeal tests are the only type of swabs that have been validated. Additionally, in the homecare setting with an interior nasal swab the FDA noted that both nostrils would need to be sampled.
Are there EUA templates for at-home tests and healthcare provider only serological tests?
· No EUA templates exist yet specifically for serological tests.
What if it has been a while and I haven’t heard back from the FDA?
· The FDA is working as fast as they can to review the EUA notifications. If their response is taking longer than you think it should, they encourage sending a follow-up email.
Once a notification is sent to the FDA under Policy D, how does the manufacturer track the progress of the notification?
· The FDA will do an immediate initial high-level review of the materials sent in the notification to ensure they meet requirements of Policy D. After that first pass, the FDA will issue an ID number (Not to be confused with an EUA number) for the device to the manufacturer. If the manufacturer does not hear from the FDA, they are able to stay on the market. Note: FDA does not issue EUA numbers for applicable tests under Policy D unless there is an EUA application submitted and approved by FDA under a different policy (i.e., A, B, C).
· Once the FDA receives the full EUA package, the FDA will review it as they have time and based on priority of the request; there is no assigned timeline for when the FDA must respond.
What if I have a non-COVID-19 product and am pursuing a traditional regulatory approval pathway. When will I hear back from the FDA on my submission?
· The FDA noted that they have all hands on deck with COVID-19 response efforts, so developers of products not related to COVID-19 may see delays in the reviews of their submissions.
How do I sign up for future townhalls with the FDA?
· Information on the town hall for COVID-19 tests can be found on the FDA website.
UserWise can assist you in swiftly obtaining Emergency Use Authorization (EUA) by providing appropriate Human Factors information to the FDA. We are experts at performing risk assessments, risk benefit analysis, and conducting user testing to confirm products can be use safety and effectively by the intended users.
We can help you obtain EUA, especially if you are pursuing an at-home test kit for COVID-19 that may require additional human factors data.
To help support manufacturers fighting COVID-19 as much as we can, we have created a knowledge base of free resources included below.
· Join our free webinar on Tuesday, April 7, 10:00 AM - 11:00 AM PST to learn more about the FDA Emergency Use Authorization (EUA) process. Register in advance to save your spot: https://userwiseconsulting.com/EUA-webinar
· Watch a short video on how we can help you navigate the EUA process: https://userwiseconsulting.com/EUA-consulting
· Book us for a free 1-hour consultation, EUA training, or Human Factors training by emailing us at userwise@userwiseconsulting.com.
︎ Lana Sneath and Denise Forkey | April 3, 2020
Test Validation
Do I need to submit for Emergency Use Authorization (EUA) for my laboratory IVD test which is a modification of an authorized test?
· The FDA does not intend to object to use of a test without a new or amended EUA; however, they expect labs to perform the appropriate validation under a bridging study.
Does data from validation tests or bridging tests need to be submitted to the FDA?
· Antibody IVD test kit developers under Policy D cannot claim authorization if they have not received EUA from the FDA. The FDA strongly encourages developers to perform a verification of the test performance; however this is not currently required.
· The FDA does not ask for the bridging testing to be submitted; however, but if you wish to share the information and share it voluntarily the FDA would like to see the data to put it on the FAQ page to help all labs.
Under Policy D, if a serological IVD test is validated for full blood, can it be used for capillary blood?
· Capillary blood can perform differently than whole blood. If the manufacturer’s instructions for use state that the sample is whole blood and the validation was performed only on full blood, the FDA suggests doing a bridging study between whole blood and capillary blood. The results of a bridging study are not required to be submitted to the FDA for review; however, the FDA is interested in reviewing this if a manufacturer volunteers to submit this data.
If my company has an antibody serological test that has been approved in the EU for some time, what do I need to do to sell it on the U.S. market?
· Notify the FDA and label it appropriately in accordance with Policy D. If you have CE mark for the IVD test kit, you may have enough data to get Emergency Use Authorization (EUA).
If one or more manufacturer or developers of antibody tests are interested in participating in a clinical trial, is there a contact office or person at the FDA they can talk to?
· FDA welcomes such studies and is happy to offer feedback. The FDA noted that they would expect to see a minimum of 30 positive patients with some IgM alone, some IgM/IgG, and ideally some IgG only. The FDA understands that given the current circumstances, the number of patients with only IgG is limited in the U.S., so they would expect it would be difficult for 30 IgG participants to be included.
· You can contact FDA at their email address to CDRH-EUA-Templates@fda.hhs.gov for further guidance.
With the serology tests, does the FDA have a source of SARS-CoV-2 positive material that IVD manufacturers could acquire for validation purposes. Is there any restriction in importing this positive material for validation?
· The FDA noted that there are overseas companies for this, but the manufacturer should assess the quality of this material. If manufacturers have trouble importing these materials, they should email the FDA.
· Additionally, the FDA is working with other agencies to help facilitate the collection and panels of immune serum for SARS-CoV-2 in the U.S. The FDA will update the FAQ page with regard to resources that could be used.
Does the FDA have any data on acceptability of saliva (nucleic acid) samples for detecting SARS-CoV-2?
· The FDA does not have policies currently issued for saliva nucleic acid tests for the detection of SARS-CoV-2 (COVID-19); however, they are interested in these tests.
· The FDA has reviewed test results for a tongue swab used in a clinic setting and the results were not promising. The FDA felt the tongue swab may not have provided enough saliva to yield promising results based on the data they have received and reviewed
· The FDA would want developers planning to sell a saliva nucleic acid test to submit a EUA amendment with the data before the FDA may authorize that test.
Labeling for Tests Under IVD Policy D
Considering EUA is not necessary for antibody testing under IVD Policy D as long as it is labeled appropriately, should manufacturers label their test kit for IVD use?
· Yes, labeling the test for IVD use is appropriate because these kits are being allowed to be marketed as IVDs under Policy D.
If I make minor updates to the labeling of my COVID-19 IVD test that falls under Policy D, do I need to notify the FDA of these changes?
· For minor changes to the labeling (e.g. extending the shelf life based), the FDA does not need to be notified. If there are changes to the performance, the FDA should be notified of the change.
At-Home Testing
What if I have a test that I want to sell for at-home use?
· The current EUA Policies for COVID-19 IVD tests do not apply to home use tests. The FDA requires IVD developers to contact the FDA to seek EUA authorization for at-home use.
· For in-home settings, the FDA is focusing on user safety, including safety of using any collection device, safety of the materials in the test device, and ensuring that results are correctly interpreted and linked back into the healthcare system. For these reasons, the FDA noted that a healthcare professional must be involved in some way to supervise the at-home testing and interpretation of the results.
· At-home use of serological tests introduces additional variables, and it is important that developers are able to demonstrate safety in these at-home environments. Blood sample collection, which is required for serological tests, is not performed routinely by lay users except for diabetics. The FDA acknowledges that the display of the results of the test devices are typically such that a healthcare provider can understand with little consideration for at-home use.
· The FDA suggested that there are clinician observation methodologies of a patient in the home that could be leveraged.
· The FDA recommends performing an observational study to identify if lay users (patients and caregivers) can use the device and instructions (under direct observation from a healthcare provider) to safely perform this testing at-home.
· The FDA also urges developers to consider how the patient will interpret the results, and how to have a clinician involved in this process. The FDA noted that some telemedicine portals are being proposed to have the user perform the test under supervision of healthcare providers to ensure the test done appropriately and results can be interpreted by a healthcare provider.
· Note: If you are looking to get your COVID-19 IVD approved for at-home use, please contact us to see how UserWise can help. We are experts in identifying use-related risks, preparing justifications for limited or remote usability testing, executing usability tests, and more (userwise@userwiseconsulting.com).
Would the FDA agree to allow for at-home use of a nasopharyngeal test? Would this need to be observed via telemedicine?
· The FDA expressed skepticism if a nasopharyngeal test could be self-administered. The FDA believes there is danger of self-harm or harm incurred by others performing this test on patients because of the invasive nature of the test. Additionally, the FDA is concerned about others helping patients collect this sample at-home because this means of collection could make patients cough or sneeze, which may spread the virus.
· Successful administration of anterior nasal swabs for nasopharyngeal tests have been demonstrated to be an effective test; however, the FDA would want to see data to prove that if a sample collected at home is returned for processing of the results, the sample is going to be in-tact and not produce false-negatives. The FDA noted that using a transportation medium that is already FDA cleared would likely not require additional testing. Otherwise, the FDA recommends performing benchtop studies (e.g. simulated shipping tests for temperature, etc.)
· Note: Foam swabs for anterior nasopharyngeal tests are the only type of swabs that have been validated. Additionally, in the homecare setting with an interior nasal swab the FDA noted that both nostrils would need to be sampled.
Are there EUA templates for at-home tests and healthcare provider only serological tests?
· No EUA templates exist yet specifically for serological tests.
Communicating with the FDA
What if it has been a while and I haven’t heard back from the FDA?
· The FDA is working as fast as they can to review the EUA notifications. If their response is taking longer than you think it should, they encourage sending a follow-up email.
Once a notification is sent to the FDA under Policy D, how does the manufacturer track the progress of the notification?
· The FDA will do an immediate initial high-level review of the materials sent in the notification to ensure they meet requirements of Policy D. After that first pass, the FDA will issue an ID number (Not to be confused with an EUA number) for the device to the manufacturer. If the manufacturer does not hear from the FDA, they are able to stay on the market. Note: FDA does not issue EUA numbers for applicable tests under Policy D unless there is an EUA application submitted and approved by FDA under a different policy (i.e., A, B, C).
· Once the FDA receives the full EUA package, the FDA will review it as they have time and based on priority of the request; there is no assigned timeline for when the FDA must respond.
What if I have a non-COVID-19 product and am pursuing a traditional regulatory approval pathway. When will I hear back from the FDA on my submission?
· The FDA noted that they have all hands on deck with COVID-19 response efforts, so developers of products not related to COVID-19 may see delays in the reviews of their submissions.
How do I sign up for future townhalls with the FDA?
· Information on the town hall for COVID-19 tests can be found on the FDA website.
How can UserWise help?
UserWise can assist you in swiftly obtaining Emergency Use Authorization (EUA) by providing appropriate Human Factors information to the FDA. We are experts at performing risk assessments, risk benefit analysis, and conducting user testing to confirm products can be use safety and effectively by the intended users.
We can help you obtain EUA, especially if you are pursuing an at-home test kit for COVID-19 that may require additional human factors data.
To help support manufacturers fighting COVID-19 as much as we can, we have created a knowledge base of free resources included below.
Free Resources
· Join our free webinar on Tuesday, April 7, 10:00 AM - 11:00 AM PST to learn more about the FDA Emergency Use Authorization (EUA) process. Register in advance to save your spot: https://userwiseconsulting.com/EUA-webinar
· Watch a short video on how we can help you navigate the EUA process: https://userwiseconsulting.com/EUA-consulting
· Book us for a free 1-hour consultation, EUA training, or Human Factors training by emailing us at userwise@userwiseconsulting.com.
︎ Lana Sneath and Denise Forkey | April 3, 2020
UserWise, LLC
Office Hours
Monday-Friday 9:00 AM - 5:00 PM PT
Copyright © 2023 UserWise, LLC. All rights reserved.