


FDA’s Dr. Feng Presents on Do’s and Don’ts for Human Factors Data in CDRH Premarket Submissions
At the recent 2019 International Symposium on Human Factors and Ergonomics in Health Care meeting in Chicago, Xin Feng, Ph.D. offered some useful tips for medical device manufacturers on how to avoid common missteps during the premarket submissions process [1]. Dr. Feng, a Human Factors Reviewer at the FDA’s Center for Devices and Radiological Health (CDRH), covered a range of issues (summarized below) that arise during the Human Factors and Usability Engineering Process.
Dr. Feng noted that, on its face, any misuse of a medical device that deviates from the intended use of the device may seem to be outside of the scope of the use-related risk analysis; however, the FDA Guidance states that manufacturers must address, “…risks arising from normal use as well as reasonably foreseeable misuse [2].” This is consistent with language presented in ISO 14971: 2007 [3], “The manufacturer shall document the intended use and reasonably foreseeable misuse.” If the misuse of a device is reasonably foreseeable or is uncovered through usability validation testing, then these cases fall within the scope of the use-related risk analysis.
In the example below Dr. Feng illustrated how to design a test scenario that provides a fuller measure of the safety of a task. This is opposed to half measures that may only uncover a user’s comprehension of certain steps, but does not probe the user’s ability to perform the task or the user’s deeper understanding of the logic behind the design of the device. In the table below, the full measure utilizes sufficiently open-ended questions that include requiring the user to explain why they would perform the task a specific way.
1. What is the Scope of Use-Related Risk Analyses Regarding the Misuse of Medical Devices?
Dr. Feng noted that, on its face, any misuse of a medical device that deviates from the intended use of the device may seem to be outside of the scope of the use-related risk analysis; however, the FDA Guidance states that manufacturers must address, “…risks arising from normal use as well as reasonably foreseeable misuse [2].” This is consistent with language presented in ISO 14971: 2007 [3], “The manufacturer shall document the intended use and reasonably foreseeable misuse.” If the misuse of a device is reasonably foreseeable or is uncovered through usability validation testing, then these cases fall within the scope of the use-related risk analysis.
2. Usability Test Scenarios: Half Measures vs Full Measures
In the example below Dr. Feng illustrated how to design a test scenario that provides a fuller measure of the safety of a task. This is opposed to half measures that may only uncover a user’s comprehension of certain steps, but does not probe the user’s ability to perform the task or the user’s deeper understanding of the logic behind the design of the device. In the table below, the full measure utilizes sufficiently open-ended questions that include requiring the user to explain why they would perform the task a specific way.

Table credit: Dr. Feng’s presentation at the HFES Healthcare Symposium slide 14
3. Failing to Understand the Limits of the Methods Used for Validation
In the example below, Dr. Feng presented a case study of the feedback the CDRH would provide when a manufacturer uses an inadequately constructed knowledge task question to test a risk control measure instead of simulated-use testing or actual-use testing. In this case the knowledge task question addresses a user’s comprehension of the statement in the Instructions for use (IFU), but does not uncover whether this knowledge is sufficient to produce the risk-mitigating behavior that would occur over multiple days.
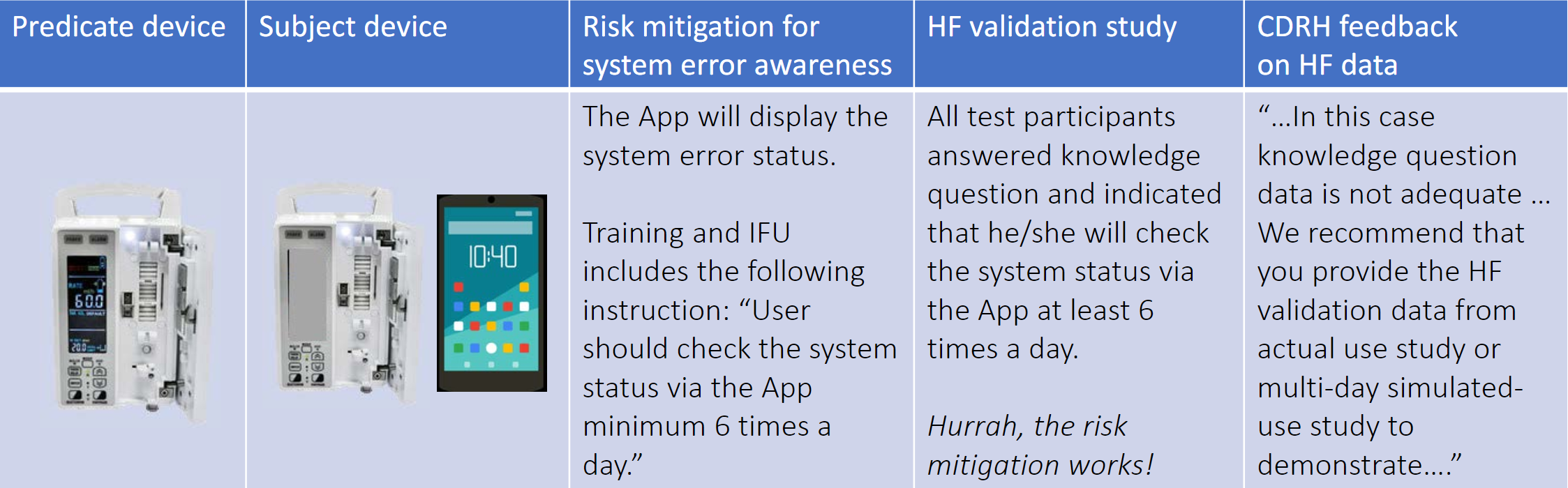 Table credit: Dr. Feng’s presentation at the HFES Healthcare Symposium slide 20
Table credit: Dr. Feng’s presentation at the HFES Healthcare Symposium slide 204. Evaluating Residual Use-Related Risk
Dr. Feng emphasized the need to refer to the criteria laid out in a device’s risk management plan when evaluating any residual use-related risks.
If the residual use-related risk is not acceptable, further risk control measures should be applied or you must provide a sound rationale that further risk control is not practicable and the benefits of the intended use outweigh the residual use-related risks.
In the example below, Dr. Feng illustrates an unsatisfactory analysis of a residual use-related risk that makes no reference to a risk management plan. In addition, it does not provide any sound rationale as to why further improvement is not practicable or why the benefits of the intended use outweigh the residual use-related risks.

Table credit: Dr. Feng’s presentation at the HFES Healthcare Symposium slide 23
5. How Should Manufacturer’s Respond to FDA-Identified Deficiencies
The final issue that Dr. Feng addressed is how manufacturers can respond to any deficiency CDRH identifies in a Human Factors Submission Package.
The FDA has issued a Guidance with a preferred format for responses to deficiencies [4]. It is best to (1) restate the deficiency and then (2) respond to the deficiency. The 3 primary options for responding to a deficiency are to provide:
- The requested information or data
- An explanation why the issue is not relevant
- Alternative information and explanation
Next Steps
If you are looking for help in preparing your human factors package for FDA submission, UserWise stands ready to help. Our competent team of consultants have years of experience successfully conducting Usability Validation Testing and preparing Human Factors Engineering Submission Reports for FDA. Contact us today to learn more about how we can assist in getting your medical product to market.
References:
1. Dr. Feng’s full slide deck.
2. FDA Guidance Document, 2017. “Design Considerations and Premarket Submission Recommendations for Interoperable Medical Devices.”
3. ISO 14971: 2007 “Application of risk management to medical Devices.”
4. FDA Guidance Document, 2017. “Developing and Responding to Deficiencies in Accordance with the Least Burdensome Provisions.”
︎ Jim Goss & Denise Forkey | May 14, 2019
Related Posts
UserWise, LLC
Office Hours
Monday-Friday 9:00 AM - 5:00 PM PT
Copyright © 2023 UserWise, LLC. All rights reserved.