


A Guide to FDA 483s and Warning Letters

FDA Inspections
As a part of the FDA regulation process, medical device manufacturers of Class II and Class III devices are subject to routine inspections of their facilities conducted by Consumer Safety Officers (CSOs) from the FDA’s Office of Regulatory Affairs (ORA). These take place at least once every two years and at even higher frequencies for newer companies in order to evaluate compliance with applicable laws and regulations regarding each product. It is crucial for manufacturers to continuously monitor all device production processes so as not to commit any regulatory violations. Otherwise, routine inspections can often lead to an FDA Form 483 observation.
What are Form 483s?
An FDA Form 483 observation (Form 483), also known as an “inspectional observation,” is a notice sent by the FDA that highlights any observed potential regulatory violations during a routine inspection. Violations can be related to all aspects of device development, including the company’s facility, product, equipment, processes and controls, or documentation records.
483s Issued in 2021
In the 2021 fiscal year alone, a total of 191 Form 483s pertaining to medical devices were issued by the FDA [1]. Of those, 64 were related to design controls (see Table 1 and Figure 1).
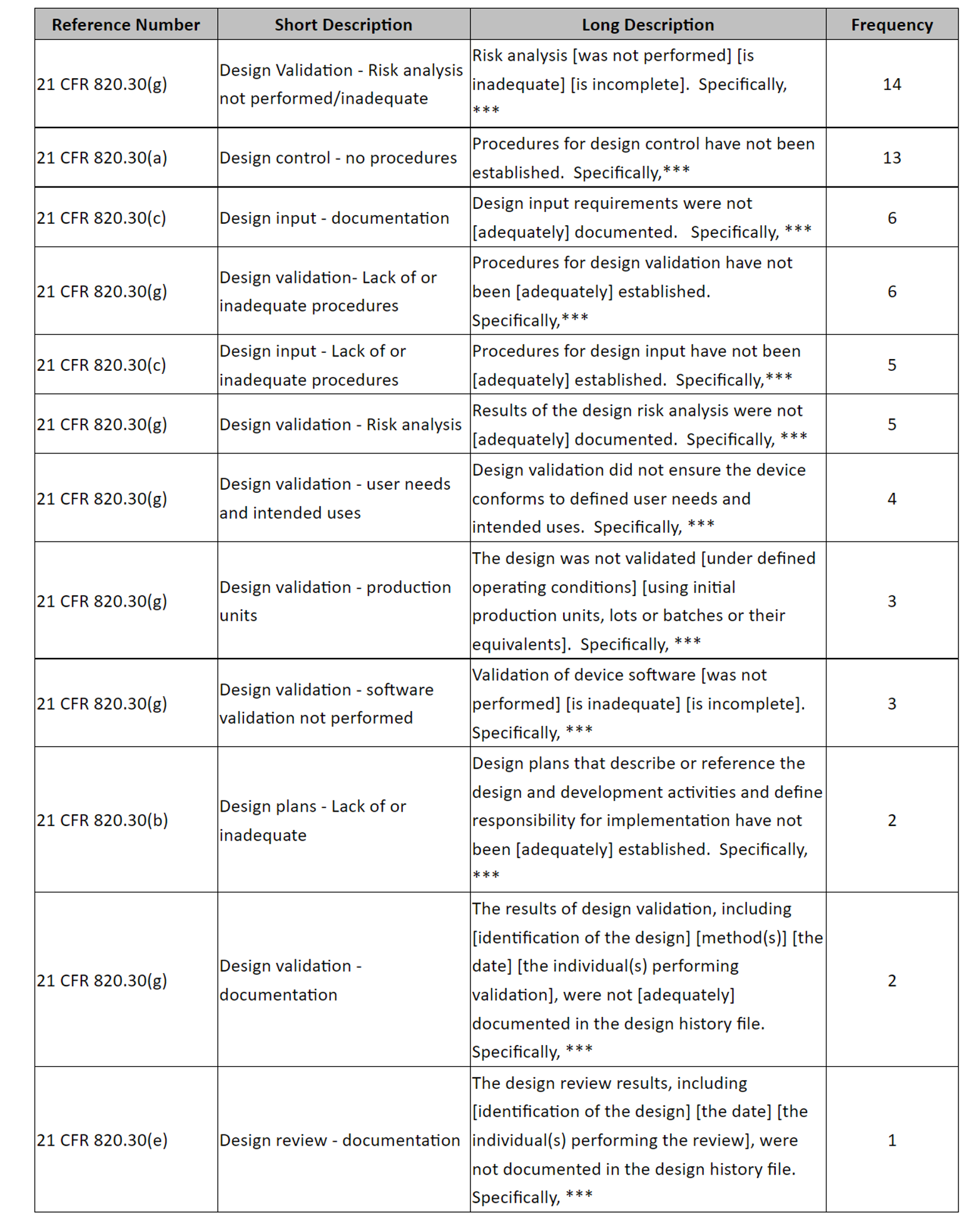
Table
1. 483s Related to Design Controls
RA: risk analysis
DI: design input
DV: design validation
DR: design review

Figure 1. Frequencies of design control violations from Table 1.
What Do I Do if I Receive a Form 483?
You’ve just received a Form 483 — now what? The first item on the checklist is to review the document with the CSO that conducted the inspection to better understand the content and have the opportunity to ask any questions. Afterwards, you will have 15 days to respond to the FDA’s concerns. It is recommended that your response letter summarizes each observation as well as your planned corrective actions.
While it is not required by law to implement corrective actions in response to observations made in a Form 483, inaction can escalate the matter to receiving a warning letter from the FDA in the future.
What is a Warning Letter and What Do I Do With it?
An FDA warning letter is a formal notification issued by more senior FDA officials after their review of the inspection report. Warning letters typically include identification of serious regulatory violations and are delivered in person. Like Form 483s, manufacturers are required to provide a response to the FDA within 15 days with planned changes.
The crucial difference between a Form 483 and a warning letter is that upon receiving a warning letter, manufacturers are required by law to implement any changes to address the FDA’s concerns.
A Closer Look at FDA Inspections
Below are two real cases of companies that received Form 483s followed by warning letters for their products:
Case 1: Manufacturer A
Manufacturer A, developer of a blood collection system, was issued a Form 483 for violation of multiple current good manufacturing requirements (cGMPs) under Title 21, Code of Federal Regulations (CFR), Part 820. Among the list were alarming observations, including manufacturing and distribution of the device with incorrect instructions for use (IFU), failure to conduct device design verification testing to support the shelf life of device accessories, and failure to establish manufacturing procedures to document and control the shelf life of components.
Although the company responded to the FDA with plans for corrective actions, they were found to be inadequate and the company was issued a warning letter 1 month later. Manufacturer A was given 15 days to provide documentation of corrections and future plans to prevent the same or similar violations from occurring again.
Case 2: Manufacturer B
Manufacturer B, developer of LED light therapy devices, was issued a Form 483 for violation of cGMPs under Title 21, CFR, Part 820 as well as major misbranding and misrepresentation. Violations of cGMPs included but were not limited to failure of establishing procedures for receiving, reviewing, and evaluation complaints regarding the device, incomplete risk analysis, and failure to maintain a device master record and device history record. Additionally, Manufacturer B published marketing statements on their website claiming that the device was FDA-approved and intended to treat neuropathy, inflammation, and other indications that were unapproved of.
Manufacturer B responded to the FDA with plans to implement procedures per requirements of Title 21 CFR, Part 820 as well as explanations for intended uses, such as the legal marketing of neuropathy treatment due to the device’s clearance to temporarily relieve pain and pain being a symptom of neuropathy. However, these were found to be inadequate and the company received a warning letter 6 months later. Manufacturer B was given 15 days to respond with documentation of steps taken to correct the violations and plans for the future.
Given these examples, it is clear that compliance to all applicable medical device standards and regulations is of utmost importance. Failure to adhere to requirements can lead to severe consequences, including product seizures, delayed regulatory approvals and clearances, and even civic penalties on top of unnecessary additional expenses. Furthermore, warning letters are publicly listed and available online, which can prove harmful to the reputation of your company to stakeholders.
How Can Your Business Prevent 483s and Warning Letters
Design control, design input documentation, and risk analysis violations prompted the most Form 483s and warning letters in 2021. Implementing human factors early in the development of your medical device is the most effective way of preventing or reducing 483s and warning letters.
Human factors can help identify the correct user group, use environment, and intended use related to your medical product. This information will be used when creating a Use-Related Risk Analysis (URRA) and will aid in identifying unknown use errors. As a result, human factors can help inform product design development and mitigate user errors related to your medical product. This process will help address concerns the FDA would indicate in a Form 483 or warning letter —ultimately saving your company time and money.
Next Steps
If you are developing a medical device, UserWise can provide the expertise you need to navigate potential regulatory hurdles. Our consultants have years of experience successfully bringing all types of medical devices to market. We can help you avoid the troubles of Form 483s and warning letters by guiding you through a seamless process ensuring compliance with all appropriate standards and regulations.
- Contact us today to set up a free 1-hour consultation for your project.
- Learn more about UserWise.
- Find out more about our unique expertise.
Subscribe to our Newsletter: Sign up for news and updates
References:
U.S. Food and Drug Administration. 2020. FDA Form 483 Frequently Asked Questions. [online] Available at: <https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/fda-form-483-frequently-asked-questions> [Accessed 25 April 2022].
U.S. Food and Drug Administration. 2022. Letters to Industry. [online] Available at: <https://www.fda.gov/medical-devices/industry-medical-devices/letters-industry> [Accessed 25 April 2022].
U.S. Food and Drug Administration. 2022. Quality System (QS) Regulation/Medical Device Good Manufacturing Practices. [online] Available at: <https://www.fda.gov/medical-devices/postmarket-requirements-devices/quality-system-qs-regulationmedical-device-good-manufacturing-practices> [Accessed 25 April 2022].
U.S. Food and Drug Administration. 2022. ORA FOIA Electronic Reading Room. [online] Available at: <https://www.fda.gov/about-fda/office-regulatory-affairs/ora-foia-electronic-reading-room?search_api_fulltext=&foia_record_type_name=483&export=yes> [Accessed 25 April 2022].
Notes:
[1] Inspectional observations for each fiscal year can be found here: <https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-observations>
︎ Amanda Yaung | May 31, 2022
Related Posts
UserWise, LLC
Office Hours
Monday-Friday 9:00 AM - 5:00 PM PT
Copyright © 2023 UserWise, LLC. All rights reserved.