


6 Ways That The Human Factors Process Differs
in Medical Devices vs Combination Products
The FDA’s Perspective on Human Factors Validation Testing and Submission

The FDA reviews both combination product and medical device submissions; however, combination products are more heavily regulated. This article will examine the different Human Factors requirements and submission strategies both types of medical products.
There are 3 types of medical products, each regulated by their own independent center at the FDA.
Background on Drugs, Devices and Biologics
There are 3 types of medical products, each regulated by their own independent center at the FDA.
Medical Products and FDA Center
| Medical Product | FDA Center | Description |
|---|---|---|
| 1. Medical Device | The Center for Devices and Radiological Health (CDRH) | Instrument, apparatus, implement, machine, contrivance, implant in vitro reagent, or other similar or related article, including a component part or accessory” that treats, prevents, or cures disease not through chemical action and is not metabolized |
| 2. Drug | Center for Drug Evaluation and Research (CDER) | Chemically synthesized agent used to treat disease or change structure or function of the body. |
| 3. Biologic | The Center for Biologic Evaluation and Research (CBER) | Isolated from a natural source (e.g., human, animal, or microorganism) and is used to treat disease or change structure or function of the body. |
Combination products are a combination of a device, drug, and/or biologic. They can be a combined product or separate products that are packaged together. For example, a combination product can consist of a medical device and a drug. During submission, any of these centers listed in the table above may review combination products depending on the Primary Mode of Action (PMOA)1. The FDA has yet to release a formal guidance regarding human factors for combination products. Currently, the FDA has released a draft guidance from 2016; however, a final version of the guidance is expected to be provided some time in 2023.
1The single mode of action of a combination product that provides the most important therapeutic action (i.e., greatest contribution to the overall therapeutic effect)
6 Ways That The Human Factors Process Differs in Medical Devices vs Combination Products
There are 6 key differences in Human Factors Validation between medical devices and combination products.
| Medical Devices | Combination Products | |
|---|---|---|
| 1. Study Participants | 15 users per user group. | 15 users per user group. Will likely need to evaluate sub-groups separately. |
| 2. Training | Only provide training when there is sufficient evidence that it will occur. | Only provide training if there is sufficient evidence that it will occur. The FDA generally expects untrained users to be evaluated for combination products. Combination product manufacturers often include both untrained and trained cohorts if training may or is likely to occur. |
| 3. Training Decay | If training is provided, a training decay of 1 hour to several days should be included. Training decay should be representative of real-world time between being trained and actually using the product for the first time. |
|
| 4. Critical Task | A task that, if performed incorrectly or not at all, would or could cause serious harm. | A task that, if performed incorrectly or not at all, would or could cause harm. |
| 5. Simulated/ Actual Use Testing | Simulated or Actual Usability Testing may be performed. Simulated Use Testing should be representative of real-world use. Simulated Use Testing is preferred due to reduced risk to Usability Study Participants. Actual Use Testing may be necessary when the medical product or the environment can affect the user’s ability to perform a critical task. |
|
| 6. Simulated Use Environment | Simulated Use Environments should be representative of real-world use environments. | |
1. Study Participants
Study Participants should be representative of users who will be using the medical product in real life.The FDA expects 15 users per user group to be evaluated during Human Factors Validation Testing. These user groups may be divided into subgroups based on age, sex, experience, education, role, or training. According to the FDA, if these sub-groups are distinct, they should be evaluated separately with 15 users per sub-group.
From UserWise’s experience, the FDA is stricter on evaluating subgroups independently for combination products, whereas for medical devices there is a better chance to justify grouping these subgroups together
Looking to recruit participants for an upcoming study?
UserWise has an in-house usability study recruitment team who can help recruit health care professional, patient, or lay user participants. Learn more.
2. Training
The FDA requires untrained participants to be evaluated if training cannot be guaranteed.Training is typically included when a training program is in place, or there is sufficient evidence that training will occur for all participants (e.g., use of a complex medical product such as robotic surgery).
If intended users may or are likely to be trained, then it is possible to include a trained cohort in addition to an untrained cohort. This is often done for submission of combination products to bolster results.
From our experience, the FDA requires untrained users for most combination products, whereas they are more likely to accept a justification for testing trained users for medical devices.
3. Training Decay
When training will be provided, the FDA expects there to be a time gap prior to testing which may range from 1 hour to several days. This time gap, known as training decay, is required for both medical devices and combination products, and should be representative of the amount of time between when a user would typically be trained on a product and when they would actually use it for the first time.For medical devices, participants are expected not to have access to any of the training material during training decay. However, for combination products, participants may be allowed to have a copy of the instructional material during the training decay period.
4. Critical Task Identification
The FDA expects that all critical tasks are evaluated as part of Human Factors Validation. The definition for a Critical Task differs between medical devices and combination products:- Critical Task for Medical Devices – Task that if performed incorrectly or not at all would or could cause serious harm.
- Critical Task for Combination Products – tasks that if performed incorrectly or not at all, would or could cause harm
5. Simulated / Actual Use Testing
When testing medical device and combination products, the studies should be representative of real-world use, with representative users performing Critical Tasks without interference from Moderators. Moderators also should perform Root Cause analysis of all use errors, difficulties, and close calls.Actual Use Testing may be conducted for either medical devices or combination products; however, it is often preferred to conduct Simulated-Use Usability Testing due to the decreased risk involved to study participants. Actual-use testing should follow the same general guidelines as simulated-use human factors validation testing. Actual-Use validation study may be necessary when the medical product or environment can affect the user’s ability to perform a critical task.
6. Simulated Use Environment
The Usability Study Environment during Simulated Use Testing should be representative of the real-world use environment. Lighting and noise are examples of environmental factors that can affect user performance. Therefore, the conditions under which simulated tasks take place and were tested represent the conditions under which users would operate the products in real life.Learn about UserWise's fully equipped Simulation Facility.
Human Factors Submission Categories for Medical Devices
In December of 2022, the FDA released a draft guidance which identified 3 submission categories for Medical Devies. These categories are identified based on the Critical Tasks, and if the submission is for a new product or a mediciation to an existing product. The category of the medical device affects submission requirements for the product.
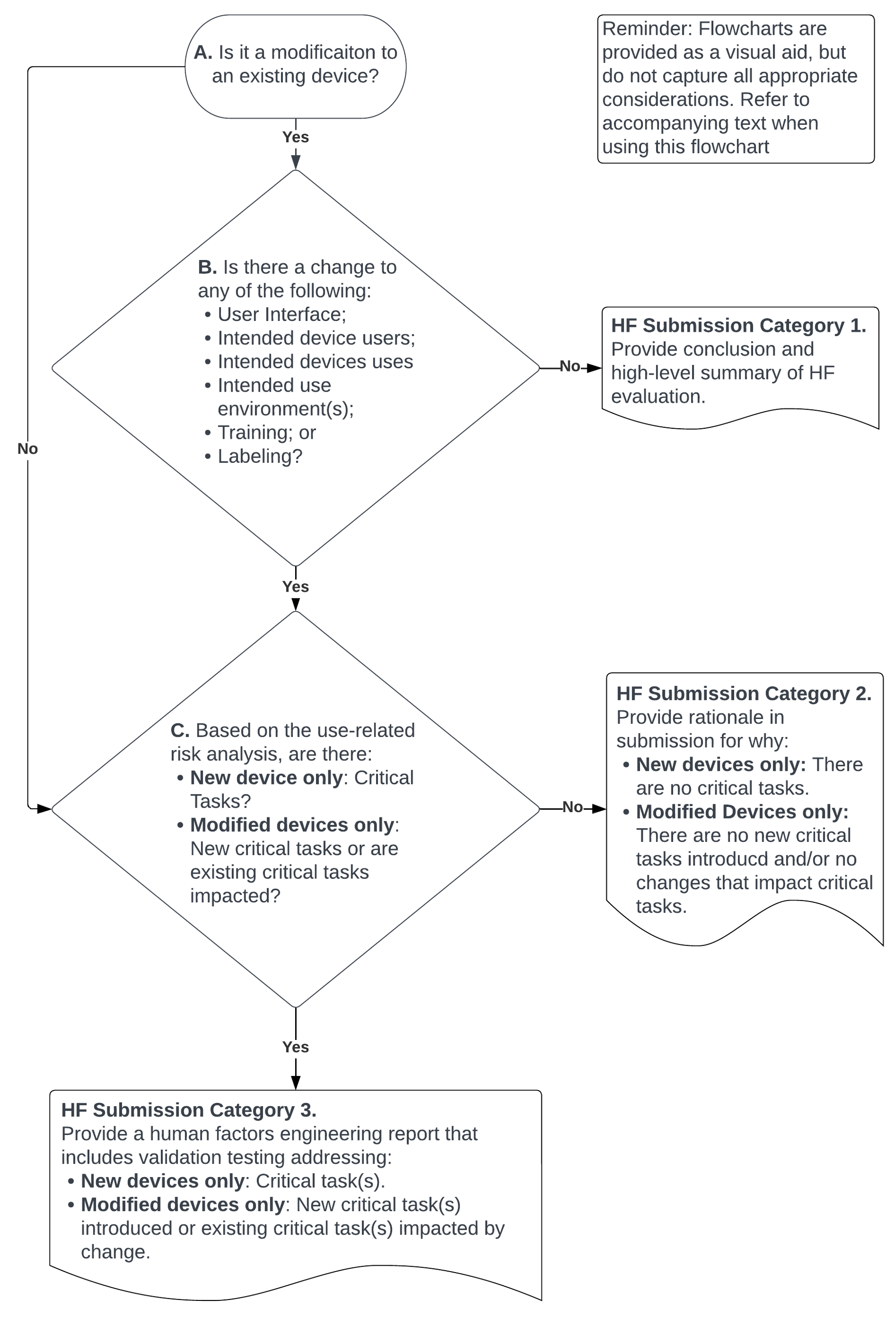
2Flowchart illustrating a risk-based approach to determine the HF Submission Category for Medical Devices.
Submission Requirements
As summarized below, there are additional submission requirements for combination products than for medical devices. If a combination product is being submitted via an abbreviated new drug application (ANDA), a Threshold analyses and a Comparative Use Human Factors Study should be conducted with the results submitted if there are any design differences between the product and the reference listed drug (RLD).
Recommended Information to Include in FDA Submission of Combination Products3
Combination Products |
|---|
|
Recommended Information to Include in FDA Submission of Medical Devices3
| Recommended Information (Report section numbers from Section V below) |
HF Submission Category |
||
|---|---|---|---|
1 |
2 |
3 |
|
| Conclusion and high-level summary (Section 1) | ︎ |
︎ |
︎ |
Descriptions of:
|
︎ |
︎ |
|
Preliminary activities
|
︎ |
||
Use-related risk analysis
|
︎ |
||
| Details of validation testing of final design (Section 8) | ︎ |
||
3Content from the December 2022 FDA Draft Guidance: Content of Human Factors Information in Medical Devices Marketing Submissions.
Conclusion
Combination products are more heavily regulated than medical devices. The PMOA determines who will govern the medical product. Regulations governing these medical products are subject to interpretation of the FDA reviewer. While there are many similarities between the requirements for medical devices and combination products, there are some differences that will affect the Human Factors Validation Testing and Submission.
Human Factors Guidance Documents
| Medical Product | Document |
|---|---|
| Medical Devices | FDA Guidance: Applying Human Factors and Usability Engineering to Medical Devices (2016) |
| Medical Devices | FDA Draft Guidance: Content of Human Factors Information in Medical Device Marketing Submissions (2022) |
| Combination Products | FDA Draft Guidance: Human Factors Studies and Related Clinical Study Considerations in Combination Product Design and Development (2016) |
| Combination Products | Human Factors Studies and Related Clinical Study Considerations in Combination Product Design and Development (draft) (2016) |
| Combination Products | Contents of a Complete Submission for Threshold Analyses and Human Factors Submissions to Drug and Biologic Applications (draft) (2018) |
| Combination Products | Principles of Premarket Pathways for Combination Products (2022) |
Next Steps
Do you need human factors guidance for your medical device or combination product? UserWise is available to help! With a proven track record consulting on more than 150 products to date, an in-house IRB and recruitment team, and contributions as a standards committee member, we are the industry’s premier human factors consultancy.
- Contact us today to set up a free 1-hour consultation for your project.
- Learn more about UserWise’s human factors services.
- Find out more about our unique expertise.
Subscribe to our Newsletter: Sign up for news and updates
︎ Franklin Li, Kaivon Assani, & Uyen Bui | January 31, 2023 [Updated April 2023]
UserWise, LLC
Office Hours
Monday-Friday 9:00 AM - 5:00 PM PT
Copyright © 2023 UserWise, LLC. All rights reserved.