


Chinese NMPA Draft Human Factors Guidance vs.
FDA 2016 Human Factors Guidance

Introduction
The National Medical Products Administration (NMPA) is the Chinese agency that regulates medical devices and drugs. In May of 2020, the NMPA issued a draft guidance document on the application of human factors and usability engineering to medical devices in China. This article provides an overview of the NMPA draft guidance, and highlights differences between the Chinese NMPA draft human factors guidance and the 2016 FDA human factors guidance.
Note: This summary is based on a translation of the draft guidance provided by Cisema as well as a translation generated using the translation feature in Microsoft Word.
Purpose of the NMPA Draft Human Factors Guidance
The Chinese NMPA draft guidance is intended to provide a framework
for the human factors design process and the necessary submission information
for medical device manufacturers. This draft human factors guidance is based on current
standards and FDA guidance:-
IEC 62366-1:2015 Medical devices – Part 1:
Application of usability engineering to medical devices.
-
IEC/TR 62366-2:2016 Medical devices – Part 2:
Guidance on the application of usability engineering to medical devices.
-
ANSI/AAMI HE75:2009/(R2018) Human factors
engineering – Design of medical devices.
- FDA, Applying Human Factors and Usability Engineering to Medical Devices – Guidance for Industry and Food and Drug Administration Staff, 2016.
Human Factors Process in the Chinese NMPA
Draft Guidance
In the Chinese NMPA draft guidance, human factors is defined as the use of knowledge around human, “anatomy, physiology, psychology, behavior, and other human factors” [2] to design medical devices with enhanced usability.
The overall human factors process outlined in the NMPA draft guidance has many parallels to the FDA human factors guidance. The Chinese NMPA draft guidance suggests that manufacturers should consider the users, user interfaces, and the use environments. These considerations align with the FDA guidance as shown in Figure 1.
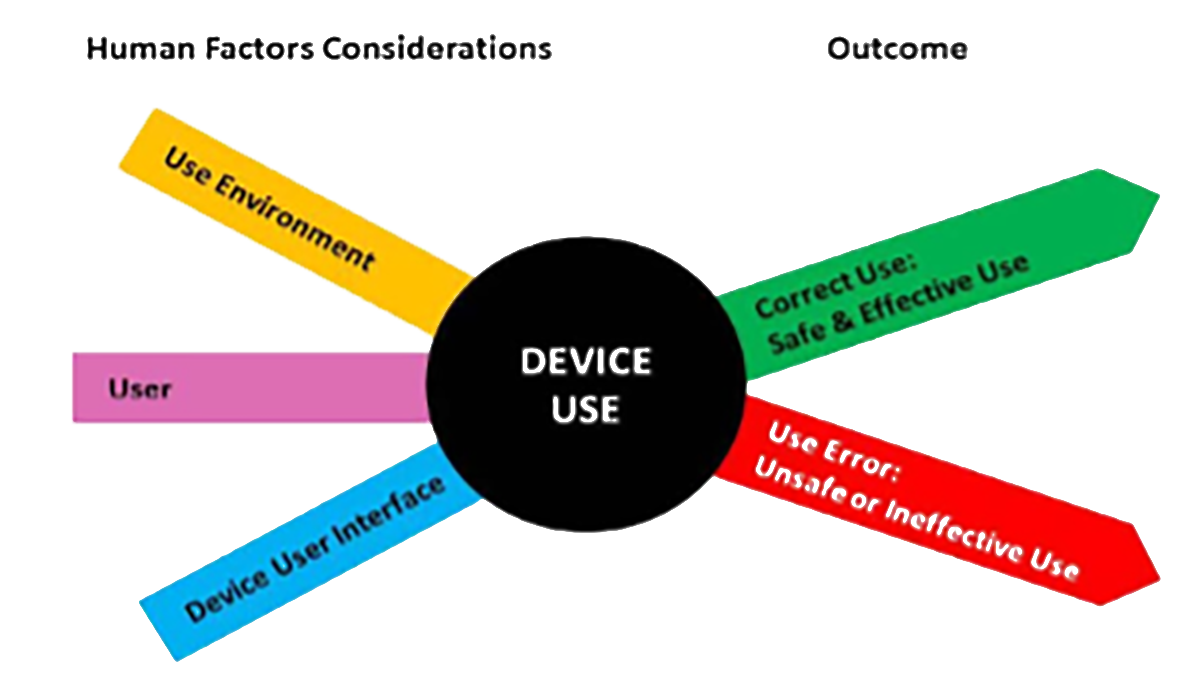 Figure 1. Human Factors Considerations from FDA Guidance [1].
Figure 1. Human Factors Considerations from FDA Guidance [1].Human Factors Tools and Methods in the NMPA Draft
Guidance
There are various human factors tools and methods that the NMPA draft guidance recommends for manufacturers to use throughout the device design process. The following tools and methods can be used to help ensure human factors engineering considerations are incorporated into the medical device design:
-
Interviews and questionnaires of the target
users or users of a similar device
-
Task analyses to determine the tasks required
for the users to operate the device and any possible risks associated with
these tasks (Failure Modes Effects Analysis (FMEA) and Fault Tree Analysis
(FTA) are recommended tools for task analysis in the Chinese NMPA draft human factors guidance)
-
Expert reviews of the design by human factors
and/or clinical experts
- Usability testing to gain feedback on potential use errors earlier in the design process
The NMPA draft guidance recommends that the specific human factors tools and methods used should be chosen to best fit the medical device being developed. Many of the tools and methods in the NMPA draft guidance are also recommended in the FDA guidance. In both guidance documents, a final summative or validation study is required once the device design is finalized to exhibit it is safe and effective before it is approved for use on the market
Four (4) Key Differences between the Draft NMPA
and FDA Human Factors Guidance
1. Device and User Scope
The NMPA draft human factors guidance states that only Class II and Class
III medical devices are in scope, whereas the FDA human factors guidance does not specifically state
which types of devices are in scope. Additionally, the NMPA draft guidance
states that personnel who install, maintain, or dispose of devices are not
currently in scope of the guidance. In contrast, the FDA guidance states that
device users can include users such as a(n) “installer, maintenance staff
member, reprocessor, [or] disposer” [1]. Therefore, there may be some medical devices
or users covered by the FDA human factors guidance that would not be in scope of the Chinese NMPA
draft human factors guidance.
2. Sample Sizes for
Usability Studies
The NMPA draft guidance has slightly different
recommendations than the FDA guidance around sample size for both formative,
and summative or validation studies. See Table
1 for specifics on the sample sizes between the two human factors guidance documents.
Table 1. Sample Size Recommendations for Human Factors Studies
| FDA Guidance Sample Size | NMPA Draft Guidance Sample Size | |
|---|---|---|
| Formative Studies | Not Specified | 5-8 users per user group |
| Summative / Validation Studies | At least 15 users per user group |
|

3. Comparative Evaluation
The Chinese NMPA draft human factors guidance contains a section on performing comparative evaluations on new and existing medical devices. Equivalent devices are defined in the draft guidance as having the same or equivalent intended use, target population, user groups, user characteristics, use environments, tasks, user interfaces, etc. The purpose of the comparative evaluation is to leverage human factors data for existing medical devices on the Chinese market when submitting for a new device. The two steps in the process of a comparative evaluation include:- Determining
what the differences are, if any, between the devices being compared
-
Gathering
information on any adverse events, recalls, or post-market problems with the
existing on-market medical device to determine if there are any new risks associated with that device that need to be considered with the new device
Table 2. Documentation Requirements for Comparative Evaluation
| No New Use Risks | New Use Risks Introduced | |
|---|---|---|
| No differences between the existing medical device and the new medical device | Documentation required:
|
Documentation required:
|
| There are differences between the existing medical device and the new medical device | Documentation required:
|
Documentation required:
|
The FDA human factors guidance recommends researching known use problems of similar devices to help guide the design process. Leveraging existing validation study data can only be done for direct modifications of an existing device under the FDA guidance.
4. Testing with Chinese Users
The Chinese NMPA draft human factors guidance states that the medical device is not guaranteed to be safe and effective in China because the use environment, users, and applications may be different in China than in other countries. Furthermore, the device may not be safe and effective when introduced in the Chinese market even if it has been proven as such in other countries.
The draft guidance recommends performing human factors validation testing in China with Chinese users unless detailed proof can be provided that the differences between countries do not impact human factors. In contrast, the FDA human factors guidance requires that the participants of human factors validation testing be U.S. residents, so this could be an important consideration for manufacturers looking to transfer an existing product from the U.S. (or another country) into the Chinese market.
NMPA Draft Guidance vs. FDA Guidance
There are many similarities between the FDA human factors guidance and the draft NMPA human factors guidance for the Chinese market. The two guidance documents largely follow the same processes to incorporate human factors concepts into the medical device design process. However, there are some differences that manufacturers should consider when taking a device into the Chinese market. Manufacturers should consider the users and use environments that may be specific to a medical device’s use in China.
Next Steps
Do you need assistance strategizing how to address the Chinese NMPA draft human factors guidance? UserWise is available to help and has expertise on multiple international human factors standards.
- Contact us today to set up a free 1-hour consultation for your project.
- Learn more about UserWise’s human factors services.
- Find out more about our unique expertise.
References:
[1] Applying Human Factors and Usability Engineering to Medical Devices - Guidance for Industry and Food and Drug Administration Staff, FDA. (February 3, 2016).
[2] Technical Review Guidelines of Human Factors Design (Draft), Cisema translation. https://www.cisema.com/wp-content/uploads/2020/06/Guideline-of-Human-Factors-Design-of-Medical-Devices-Draft-EN.pdf
Subscribe to our Newsletter: Sign up for news and updates
︎ AnnieRuth Sawyer | July 27, 2022
UserWise, LLC
Office Hours
Monday-Friday 9:00 AM - 5:00 PM PT
Copyright © 2023 UserWise, LLC. All rights reserved.